KD (also called mucocutaneous lymph node syndrome) is one of the most common vasculitides of childhood. The incidence of KD is greatest in children who live in East Asia (e.g., Japan, Korea, Taiwan) or are of Asian ancestry living in other parts of the world. Other risk factors include male sex, age between six months and five years, and family history of KD. KD occurs only rarely in adults.
KD is typically a self-limited condition, with fever and manifestations of acute inflammation lasting for an average of 12 days without therapy. Still, cardiac complications can lead to significant morbidity and mortality. Timely diagnosis and initiation of appropriate therapy minimize cardiac sequelae and improve clinical outcomes.
The etiology of KD remains unknown. Inflammatory cell infiltration into KD vascular tissue leads to vascular damage, but the inflammatory infiltration stimulus has not been identified.
The similarities between KD and other pediatric infectious conditions suggest that a transmissible agent causes KD. However, no studies have convincingly identified a specific virus, bacterium, or bacterial toxin, or other pathogen-associated with KD. The etiology may be a previously unidentified infectious agent. An alternative theory to a specific inciting agent is that KD represents a final common pathway of immune-mediated vascular inflammation following various inciting infections or environmental antigens.
Genetic factors appear to contribute to this disorder's pathogenesis, as suggested by the increased frequency of the disease in Asian and Asian-American populations and family members of an index case. Several gene polymorphisms are associated with increased susceptibility to KD, and some of these variants are also associated with coronary artery lesions and aneurysm formation.
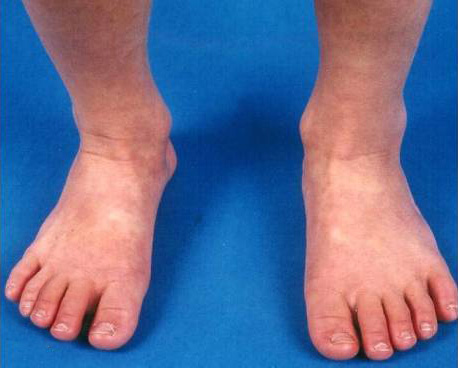
KD is characterized by systemic inflammation manifested by fever and mucocutaneous involvement, including:
- Bilateral nonexudative conjunctivitis
- Cervical lymphadenopathy
- Erythema of the lips and oral mucosa
- Extremity changes
- Rash
These findings are the basis for the diagnostic criteria for KD. Patients who lack a sufficient number of findings to fulfill the classic criteria may have incomplete KD.
These findings are often not present at the same time. Thus, repeated histories and physical examinations are important in making a timely diagnosis of KD in children with fever and signs of mucocutaneous inflammation.
Infants and possibly adults are more likely to present with incomplete KD. Infants are at greater risk for cardiovascular sequelae, possibly due to a delay in diagnosis and intervention. Thus, infants six months of age or less with unexplained fever for at least seven days should be evaluated for KD, even if they have no clinical findings of KD.
No laboratory studies are included among the diagnostic criteria for complete KD. But certain findings characteristic of KD may support the diagnosis in ambiguous cases. However, the presence of compatible laboratory features strongly supports the diagnosis.
According to classical criteria, the diagnosis of KD requires the presence of fever ≥five days, combined with at least four of the other five signs of mucocutaneous inflammation, without any other explanation. A significant proportion of children with KD have a concurrent infection. Therefore, ascribing the fever to such an infection or KD requires clinical judgment.
Additional clinical and laboratory features are often used to guide diagnosis in children who have fewer than five KD criteria (incomplete KD). The criteria to diagnose incomplete KD and initiate treatment in a child in whom an experienced clinician believes that KD is the probable diagnosis include elevated CRP or ESR and three or more abnormal supplemental laboratory findings OR abnormal echocardiography. A child with incomplete KD whose diagnosis is delayed is more likely to develop CA abnormalities. Thus, KD should be in the differential diagnosis of any child with unexplained fever lasting five or more days.
KD is most commonly confused with infectious exanthems of childhood. The presence of clinical features not commonly found in KD, including exudative conjunctivitis, exudative pharyngitis, discrete intraoral lesions, bullous or vesicular rash, splenomegaly, or generalized lymphadenopathy, suggest another diagnosis. Nonetheless, KD is sufficiently pleomorphic that none of these findings can definitively exclude the diagnosis. Children with KD can have concurrent infections, particularly with viruses circulating in the community at their diagnosis.
Patients who fulfill the criteria for complete KD or incomplete KD require treatment because of the risk of cardiovascular complications that may result in significant morbidity and even mortality.
A single IVIG dose (2 g/kg administered over 8 to 12 hours) is recommended in patients with KD. IVIG is most effective if administered within the first ten days of illness before aneurysms typically develop. Nonetheless, IVIG should be administered even beyond this 10-day window in patients with evidence of persistent vasculitis or systemic inflammation (e.g., persistent fever, elevated acute-phase reactants).
In patients with KD, aspirin should be administered during the acute phase of the illness. The AAP and AHA have recommended a broad range of aspirin doses (30 to 100 mg/kg/day), but it is not clear that any amount of aspirin has long-term benefits. A total daily aspirin dose of 30 to 50 mg/kg/day in four divided doses (maximum dose 4 g per day) is advised. Still, it should be held for any contraindication, particularly exposure to varicella or influenza. The aspirin dose is decreased to 3 to 5 mg/kg/day 48 hours after fever resolution. Aspirin is continued until laboratory markers of ongoing inflammation (e.g., platelet count and ESR) return to normal unless CA abnormalities are detected by echocardiography, in which case aspirin therapy is continued.
For Japanese children, well-validated criteria, such as the Kobayashi criteria or similar, can be used to select patients at increased risk of IVIG resistance. Such high-risk patients are most likely to benefit from adjuvant treatment with glucocorticoids in addition to IVIG.
For non-Japanese patients, the criteria for identifying at-risk children are not well validated. In these patients, the following may be used to select those who are most likely to benefit from augmented treatment with glucocorticoids in addition to initial IVIG:
- Age ≤12 months (and particularly age <6 months)
- Enlarged CAs at presentation (before IVIG treatment)
- KD associated with shock
- KD presenting with MAS
In children with KD who are determined to be at increased risk of IVIG resistance, glucocorticoids should be added to initial IVIG therapy. A total of 15 days of prednisone/prednisolone (five days each of 2 mg/kg, 1 mg/kg, and 0.5 mg/kg), starting with an intravenous formulation and switching to oral glucocorticoids 12 to 24 hours before expected discharge, is suggested. A reasonable alternative, particularly in non-Japanese patients, is to treat with IVIG and aspirin alone since most children with KD recover with standard therapy and have a low risk of CA aneurysms or other complications.
Prognosis is based upon the severity of CA involvement as a marker of risk for MI. After the baseline echocardiogram is obtained at diagnosis, echocardiography is usually repeated at approximately two and six weeks of illness to evaluate CA involvement. Examinations and echocardiograms are repeated more frequently in children with CA abnormalities. Children also should be monitored carefully for any signs of persistent illness during the first two weeks after treatment, as those with ongoing inflammation, particularly fever of any degree, are at the highest risk of developing CA abnormalities.
The AHA and AAP have developed guidelines for subsequent therapy, physical activity, and follow-up visits (schedule and content) based on MI's relative risk.
Patients without any cardiovascular abnormalities appear to be clinically healthy at long-term follow-up (range, 10 to 21 years). However, it is unknown whether they are at increased risk for atherosclerotic heart disease.
Administration of live-virus vaccines (e.g., measles, varicella) for at least 11 months in children who have been treated with IVIG should be postponed because passively acquired antibodies can interfere with vaccine immunogenicity. One exception to this postponement is in children residing in communities experiencing an outbreak of a vaccine-preventable disease. Another exception is children on long-term aspirin therapy. Children who are ≥12 months of age and are on long-term, moderate- or high-dose aspirin therapy (30 to 100 mg/kg/day) should receive the varicella vaccine because of the increased risk of Reye syndrome. In situations where vaccination is not postponed, the vaccine should be repeated at least 11 months after administering IVIG. Influenza immunization, recommended in all children over six months of age, is also particularly important in those who require long-term high-dose aspirin therapy because of the possible increased risk of Reye syndrome.
Some patients with KD have persistent or recurrent fever despite treatment with IVIG and aspirin. Prolonged fever is a risk factor for CA sequelae in KD. In these patients, other causes of fever should be considered. Risk factors for failing to respond to a single dose of IVIG include:
- Age <6 months
- Elevated CRP
- Low platelet levels
- Low sodium
- Male sex
Patients with refractory KD are at increased risk of developing CA aneurysms.
Additional therapy is recommended in children with KD who have persistent fever 24 hours after completing initial therapy or whose fever returns after an afebrile period (up to two weeks after starting treatment). Retreatment is suggested with a single dose of IVIG (2 g/kg). Additional dose(s) of IVIG may be administered before considering other medications in those who remain febrile. However, the benefits of a total dose of more than 4 g/kg of IVIG have not been demonstrated.
For patients who do not respond to retreatment with IVIG, treatment with glucocorticoids is suggested. Pulsed-dose methylprednisolone (30 mg/kg per day administered on one to three consecutive days) once daily until a full response (e.g., resolution of fever) is obtained or the patient has received three doses is suggested. A 15- to 30-day course of twice or thrice daily oral prednisone (total daily dose 1 to 2 mg/kg) may be equally or more effective than IV methylprednisolone in children with KD who are resistant to IVIG.
Therapeutic alternatives, which have demonstrated effectiveness for other vasculitis forms, include TNF-alpha inhibitors, other immunosuppressive agents, and plasmapheresis. However, there are limited data concerning these agents' risks and benefits for refractory KD and only a few small trials to evaluate them. The most commonly used of these agents is infliximab, a monoclonal antibody against TNF-alpha. In multiple trials, it effectively lowers fever, laboratory markers of inflammation, and signs of mucocutaneous inflammation, but there is minimal evidence that it improves CA outcomes. Infliximab (5 mg/kg) is typically given to children who have evidence of active vasculitis despite receiving two IVIG courses (total 4 mg/kg) and one to three IV doses of methylprednisolone.