Only Advanced Practice Nurses will receive credit for this course.
Listen to Audio
Get one year unlimited nursing CEUs $39Sign up now
This peer reviewed course is applicable for the following professions:
Advanced Practice Registered Nurse (APRN), Certified Registered Nurse Anesthetist (CRNA), Clinical Nurse Specialist (CNS), Respiratory Care Practitioner, Respiratory Therapist (RT)
This course will be updated or discontinued on or before Monday, October 18, 2027
Nationally Accredited
CEUFast, Inc. is accredited as a provider of nursing continuing professional development by the American Nurses Credentialing Center's Commission on Accreditation. ANCC Provider number #P0274.
Outcomes
Acute Respiratory Distress (ARDS) clinical presentation is marked by disruptions to the alveolar-capillary complex mediated by inflammation, interalveolar edema primarily caused by protein-rich fluids influx, reduced alveolar clearance, and increased pulmonary resistance. Beyond sepsis-induced inflammation, a mechanical ventilation regimen can also propagate ventilator-related injury that may precipitate ARDS. The mechanisms involved in the pathophysiology of ARDS are complex and are primarily under clinical investigation. However, the symptomatology, management guidelines, and postoperative rehabilitation plans for this condition have been mapped out. This course is designed to evaluate ARDS in perspective, discussing its clinically-accepted definitions and modifications made in subsequent years. We have created a detailed outline of the pathophysiology, epidemiology, diagnosis criteria, clinical management, and rehabilitation care guidelines. The different mediators responsible for Diffuse Alveolar Damage (DAD), the hallmark of ARDS, will also be discussed based on available clinical evidence.
Objectives
After completing this continuing education course, the participants will be able to meet the following objectives:
Summarize the pathophysiology of non-HF ARDS.
Describe the clinical presentation for non-HF ARDS.
List the criteria needed for the diagnosis of non-HF ARDS.
Explain the management guidelines of non-HF ARDS.
Identify the postoperative and rehabilitative care for non-HF ARDS.
CEUFast Inc. and the course planning team for this educational activity do not have any relevant financial relationship(s) to disclose with ineligible companies whose primary business is producing, marketing, selling, re-selling, or distributing healthcare products used by or on patients.
Last Updated:
$39 Unlimited Access for 1 Year (Includes all state required Nursing CEs)
No Tests Required (Accepted by most states & professions)
Nursing Assistants from California, only. You must read the material on this page before you can take the test. The California Department of Public Health, Training Program Review Unit has determined that is the only way to prove that you actually spent the time to read the course. Less
Medical conditions characteristic of respiratory dysfunctions can be lethal as they disrupt the arterial oxygenation level and impair the functional supply of oxygen to major organs. First case clinical study records described the development of severe or mild respiratory distress in patients with severe pancreatitis, sepsis, nonthoracic injuries, massive transfusion, and other conditions. Bouts of respiratory distress were reported in many patients during inpatient admissions and, sometimes, a few hours or days after discharge. To better explain the symptoms and presentations of the condition, the term acute respiratory distress syndrome (ARDS) was coined as descriptive medical terminology for the type of respiratory distress. As with other medical conditions, research and clinical studies unveiled new information about this condition, including reports of a similar presenting group of symptoms in children.
Olusola Cristiano, a 45-year-old Ghanaian immigrant, had earned an industry reputation for a rigorous work schedule and on-time project delivery. His recent work had him miss a few hours of sleep as he traveled across different time zones on business trips. In January, Olusola presented to the emergency department of the company's insurance provider with complaints of cough, chills, and severe, pulsating right-sided chest pain. He explained that the pain had lasted for one month, causing mild difficulties in breathing. He had no episodes of vomiting, muscle cramps, or myalgias.
His medical history reported a long-term childhood exposure to a relative diagnosed with Tuberculosis. In the last two months, he had lost approximately 35 pounds and had several episodes of nightly sweats. He denied recreational drug use but had smoked at least two packs of cigarettes weekly for the past two years. Before venturing into the construction industry, he had worked previously in a steel mill, collecting coins and cleaning them with mercury sprays. He is a known alcoholic, not currently on any medication, and has no clinical record of comorbidities.
On examination, Olusola's pain seemed pleuritic with an associated cough productive of yellowish, spotted sputum without hemoptysis. The attending resident admitted him into the General Observatory to treat community-acquired pneumonia. He was started on a prescription order for Azithromycin, Ceftriaxone, and oral pain relievers. The initial therapy plan was to manage him as an inpatient and recommend discharge once his condition improved. On the third day, Olusola reported improved pain control in the chest but complained of mild difficulties in breathing. On day 5 of hospital admission, the attending resident was urgently alerted to his respiratory status worsening.
He was transferred to the ICU for unexplained ARDS. On admission to the ICU, Olusola appeared dyspneic. On auscultation, breath sounds were diminished on the right side in the upper zone without the presence of adventitious sounds. The white blood cell count was 28 cells/µl, and the red blood cell count was 51 cells/µl. His blood pressure was 165/95 mmHg, heart rate was 131/min, respiratory rate was 32/min, and body temperature was 38.2 degrees Celsius. His oxygen saturation on room air was 77% and 90% on 40% venti-mask. He was ventilated and was on synchronized intermittent mandatory ventilation mode. Positive end-expiratory pressure (PEEP) was increased to 12 cm of H2O, the respiratory target was set at 20 breaths per minute, and the inspiration rate was 1:2. He was in a prone position and maintained with 0.9% normal saline at 40ml/hour.
On the subsequent evaluation round, his blood pressure dropped to 125/85 mmHg, and his lung function ratio improved from 192 to 300 and finally to 400. His breathing pattern had improved significantly, and arterial oxygen saturation had increased to 98%. The consultant recommended gradual oxygen weaning and adaptive support ventilation (ASV). The discharge order recommended 24-hour stabilized vitals before discharge can be considered.
In light of new clinical information, in 1994, the American-European Consensus Conference (AECC) developed a clear definition of this condition, ultimately renaming it (ARDS). ARDS was an acute condition characterized by bilateral pulmonary infiltrates and severe hypoxemia. In 2011, this definition was modified by the Berlin Definition of ARDS. ARDS has been redefined as a condition lasting at least one week with bilateral opacities and edema. These opacities are not fully explained by effusions, consolidation, or atelectasis (radiographic examination), and the edema is not explained by fluid overload, cardiac failure, or respiratory severity.
Subsequently, the Berlin definition classified ARDS into three distinct categories differentiated by the severity of oxygenation compromise. Unlike the AECC's previous definition, it excluded acute lung injury (ALI) from the clinical characteristics of ARDS, removed the requirement for wedge pressure <18, and included the requirement of PEEP. The three categories identified were mild (PaO2/ FiO2 200-300), moderate (PaO2/ FiO2 100-200), and severe (PaO2/ FiO2 ≤100) (Matthay et al., 2021). Objective assessments, including echocardiogram, were recommended as diagnostics methods if risk factors for ARDS were absent. As with many medical conditions, basing the diagnosis of ARDS on clinical criteria was necessary as it is practically impossible to obtain direct measurements of lung injury by pathological samples of lung tissues in most patients.
Study surveys reported an increased number of respiratory distress symptoms in the pediatric population, highlighting the need for a harmonized ARDS diagnosis and management protocol in this population. Protocols and other clinical specifics were developed at the Pediatric Acute Lung Injury Consensus Conference (PALICC).
Specific terms developed from this include:
Oxygenation index (OI)
Oxygen saturation index (OSI)
Pulse oximetric saturation to the fraction of inspired oxygen ratio (S/F, SPO2/FiO2)
The following was recommended for pediatric patients:
The utilization of low tidal volume (5-8 mL/kg of predicted body weight)
Limiting plateau pressure to 28-32 cm H2O
Permissive hypercapnia strategy
PEEP in the range of 5-15 cm H2O
Acceptance of low SPO2 in the range of 88-92% if PEEP is as high as 10 cm H2O
The pathophysiology of this condition is challenging to pinpoint as the etiology is multifactorial. In the early research describing the pathophysiology of ARDS, a direct pulmonary or indirect extrapulmonary insult was implicated in the proliferation of inflammatory mediators promoting neutrophil accumulation in the microcirculation of the lung. With the current research on this condition, etiologies that trigger this syndrome have been grouped into direct and indirect causes. Direct causes trigger primary injuries to the lung epithelium and include pneumonia, aspiration, drowning, and toxic inhalation. Indirect causes, however, trigger diffuse damage to the lung's vascular endothelium and cause systemic lung inflammation. These triggers include:
Extrapulmonary sepsis
Non-cardiogenic shock
Pancreatitis
Adverse drug reactions
Vasculitis
Trauma
Transfusions
The histological hallmark of ARDS appears to be diffuse alveolar damage (DAD). These etiologies provoke a systemic inflammatory response in the lung.
Various pathways, mediators, and molecular systems contribute to altered alveolar endothelial and epithelium porosity. An adherens junctional protein, the vascular endothelial cadherin (VE-cadherin, has been identified to perfume critical physiological functions in maintaining barrier integrity in the lung microvessels. Disruption of the homophilic bonds on the VE-cadherin destabilizes lung barrier integrity. Antibodies against VE-cadherin and other destabilizing agonists such as thrombin, TNF, VEGF, and leucocyte signals have disrupted VE-cadherin bonds, prompting lung edema (Lupu et al., 2020). In sharp contrast, factors that stabilize VE-cadherin bonds, including genetic manipulation of VE-cadherin-catenin interactions or preventing the dissociation of a phosphatase group from VE-cadherin, reduce the influx of leucocytes and fluids into the alveolar space. The interaction has been proven in clinical studies exploring potential treatment options for ARDS in mice (Jones et al., 2022).
Recent studies on a new pathway implicated in alveolar epithelium damage focus on the role of a neutrophil-platelet complex. Neurons in the intravascular and extravascular compartments often form a complex with platelets. These platelets have intricate thrombo-inflammatory activities and, most importantly, the ability to deploy neutrophil extracellular traps (NETs). Histological researchers describe NETs as filamentous chromatin fiber complexed with neutrophil-derived antimicrobial proteins. These traps reportedly evolved as an innate mechanism for pathogen containment and clearance. However, they are also involved in inflammatory insults to major organs, including the lung (Lefrancais et al., 2018). In experimental models, NETs correlate with alveolar-capillary and epithelial disruptions causing alveolar edema and damage. Research observations have also suggested an early intravascular interaction of platelets with monocytes in an aggregation mechanism similar to the neutrophil-platelet association. These aggregations involving monocytes also reportedly drive the development of ARDS in patients at risk. The pathway involved in recruiting neutrophils and monocytes to the lung is influenced by intra-alveolar macrophages releasing chemotactic factors. These factors, including CC-Chemokine ligand 2 and IL-8, enhance neutrophil recruitment as a physiological response to acute pulmonary infections (Abdulnour et al., 2018).
Studies have shown that the balance between the protective and harmful innate and adaptive immune responses may also determine the severity of alveolar injuries. Acute inflammatory responses to pathogens and toxins in disease cycles of lung infections can precipitate ALI. These inflammatory responses release leucocyte protease, synthesis of chemokines and cytokines, toll-like receptor engagement, and generation of reactive oxygen species (Xie et al., 2021). Research submissions have also highlighted how molecular events governing the balance between angiotensin-converting enzymes 1 and 2 (ACE I and II) may also contribute to inflammatory lung injuries as a direct consequence of viral infection, acute injury, and sepsis (Wu et al., 2022). Injuries cause airspace and interstitium edema by developing a protein-rich neutrophilic exudate. The development of exudate compromises gas exchange and reduces the integrity of the normal respiratory cycle. Understanding the pathophysiology of ARDS regarding alveolar integrity involves studying the normal gas transfer flow of the lung.
A healthy lung is physiologically designed to exchange carbon dioxide and oxygen across the distal alveoli-capillary unit. The selectively permeable barrier establishing a gaseous exchange in the lung is formed by a single layer of the endothelial lining, aggregated together by tight junctions and plasma membrane structures such as adherens (Chen et al., 2021). The surface of this expansive boundary is lined by fat alveolar type 1 cells (ATI) and cuboid-shaped alveolar type 2 cells (ATII), forming a barrier restricting the free flow of solutes but allowing the diffusion of oxygen and carbon dioxide. Surfactants secreted by the Alveolar cells serve critical functions in the reduction of surface tension and in maintaining active gas exchange by keeping the alveoli complex open. Sodium channels and basolateral Na+/K+ -ATPase pumps on the alveolar cells help absorb excess fluid from the airspaces by vectorial ion support (Esquivel-Ruiz et al., 2021).
With this mechanism, there is the reabsorption of edematous fluid in the event of alveolar edema. Fluid reabsorption through this mechanism into the lung is primarily transported and removed by lung microcirculation and the lymphatic network. In addition to the tight barrier structure, the cellular makeup of a normal, healthy alveolus also includes alveolar macrophages ready for rapid deployment into the lung microcirculation. These macrophages and a cellular population of platelets, monocytes, and neutrophils control the defensive response of the healthy alveolus and have a critical function in the pathophysiology of ALI.
Image 1: Structure of an Alveolus
In ARDS, the lung epithelium and the selective barrier on the alveoli surface are compromised, forcing liquid and protein across the endothelium. The absorption of protein and fluids across this barrier precipitates edema in the lung interstitium (Meyer et al., 2021). From the lung interstitium, fluid flows to the alveoli, taking advantage of damage to the alveolar epithelium. Increased alveolar-capillary permeability to protein, neutrophils, and blood cells results in the accumulation of fluids in the alveoli (Doig et al., 2022). Research studies have identified the impaired excretion of carbon dioxide due to alveolar-capillary compromise as the root component of respiratory failure in ARDS. Impaired carbon dioxide excretion results in elevated ventilation associated with a greater volume of breath participating in carbon dioxide excretion (an increase in pulmonary dead space). In addition to increased pulmonary dead space, a decrease in respiratory compliance is another independent predictor of mortality in ARDS (Patel et al., 2022).
The expansive pathophysiology of ARDS also includes eosinophilic depositions called hyaline membranes. Hyaline membranes are a defining feature of DAD (Cardinal-Fernandez et al., 2017). Other features described so far include fibrin deposition, alveolar hemorrhage, alveolar atelectasis, and accumulation of white blood cells. The acute exudative phase of ARDS lasts approximately 7 days and usually resolves completely. In some cases, residual pulmonary fibrosis may develop, with the alveolar space filled with mesenchymal cells and new blood vessels. With recent research on DAD, pulmonary fibrosis appears to be facilitated by interleukin 1 (IL-1). Pathological progression to the fibrosis stage may be predicted early by investigating the possibilities of increased procollagen peptide III (PCP-III) levels in the fluid obtained by bronchoalveolar lavage (BAL) and lung biopsy specimens (Davidson et al., 2020).
Hyperplasia of the alveolar cells follows in a proliferative phase. The proliferative phase usually lasts for about 3 weeks. Analyses of the lungs of patients dying from oxygen toxicity formed the basis for the original description of DAD phases in ARDS. However, recent clinical evidence identified similar histological changes in the lungs of patients with multiple other underlying conditions (Thille et al., 2017). In a post-mortem analysis of lung samples collected over two decades (1992-2010), researchers reported that 45% of patients who met the Berlin criteria for ARDS had DAD as the underlying pathology for alveolar dysfunction. Of the study population, 55% had pneumonia, with the infiltration of neutrophils in the alveoli and distal bronchioles as the underlying trigger for ARDS. Another meta-analysis of open lung biopsy samples in ARDS patients provides supporting evidence for the decade-long study. One study reported that DAD was only present in 48% of ARDS patients in their systematic review study and was associated with a higher mortality index (Kao et al., 2017). The severity of hypoxemia and the sequential organ failure assessment appears to vary with no significant pattern in patients with or without DAD. The other bulky half of the population presented with various ARDS triggers.
Early histology studies on ARDS identified the association of DAD with the severity of ARDS, with the acute exudative phase presenting the most viable window for therapeutic intervention. Electron microscopic analysis identifies multiple alterations in the physiological composition of the epithelia and endothelial cells (Ortiz et al., 2019). Focal epithelial destruction and denudation of the alveolar basement membrane have been identified in the early involvement of ATI cells. However, ultrastructural analysis of samples could not conclusively show abnormalities in the normal alveolar barrier structures responsible for regulating gas exchange, protein influx, and fluid movement across the lung capillaries. In many cases, the alveolar endothelial cells may be preserved morphologically with the continuity of the endothelial lining maintained. Studies have shown extensive epithelial necrosis in the exudative phase before mesenchymal cell invasion, with suggestive evidence for apoptosis (Hussain et al., 2021). The release of pro-coagulant factors and subsequent deposition of fibrin during the exudative phase appears to be triggered by injured but intact alveolar epithelial cells. Pro-coagulation factors and fibrin are also deposited adjacent to injured endothelial cells in the alveoli (Kaku et al., 2020).
Although the complete mechanism of alveolar damage in ARDS remains under investigation, there are studies implicating apoptosis, pyroptosis, and other alternative cell death pathways. These occurrences are in addition to early submissions on the roles of increased lung vascular permeability and endothelial junction breakdown in the pathophysiology of ARDS. In the damaged alveoli, neutrophil deposition occurs due to endothelial activation. The neutrophil-platelet aggregates play a synergistic role in increasing lung vascular permeability to protein, blood cells, and fluid. Increased vascular permeability significantly increases the frequency and risk of hypovolemia and multiple organ failure. In autopsy analysis of damaged alveoli, the epithelial cell disruptions are significantly pronounced compared to ultrastructural alteration of alveolar endothelial cells (Kersten et al., 2020). Multiple autopsy studies have also implicated pathogens and barrier-destabilizing factors as prominent causes of alveolar endothelial disruption. Others include:
Circulating leukocytes and platelets
Pro-inflammatory signaling molecules such as tumor necrosis factor (TNF)
Therapeutic interventions and early experimental therapy models focused on mitigating endothelial permeability and reducing excessive leukocyte accumulation by re-establishing the physiological integrity of the alveolar endothelial junction. Early research on this hypothesis explored the genetic replacement of VE-cadherin with a fusion construct that prevents its internalization. Research results suggest a significantly reduced alveolar neutrophil accumulation in LPS-challenged mice. The alveolar permeability index was also significantly reduced. In an experimental simulation of ALI, the alveolar epithelium showed more resistance to injury than the endothelium. However, injuries to both structures include the disruption of intercellular junctions. In most cases, the extent of epithelial injury directly describes the severity of ARDS.
Clinical recovery and symptom resolution of ARDS can be achieved with the repair of the injured alveolar epithelium. On average, the time frame for epithelial repair may be three weeks or several weeks, depending on the severity of the alveolar compromise (Beretta et al., 2021). Generating new ATI cells is critical to alveolar repair since these cell types comprise about 95% of the alveolar epithelium surface area. Beyond the structural composition, ATI cells play principal roles in gas exchange across the alveolar surface. In many cases, ATII cells proliferate to provide a provisional epithelial barrier before transdifferentiating into ATI cells. Alternate progenitor cells, including club cells, keratin-5- expressing cells (KRT5+), and bronchoalveolar stem cells, can be mobilized to create new alveolar epithelial cells through proliferation. These alternative proliferation pathways are complementary options to ATII cells for alveolar repair.
The biological process and drivers for differentiating ATII cells from ATI cells are not entirely understood. However, recent research focuses on the alternate progenitor cell and default cell cycle for ATI cells, suggesting the principal role of deactivating WNT–β-catenin (Liberti et al., 2021). In addition to WNT–β-catenin deactivation, hypoxia-inducible factor (HIF) and notch signaling drive ATII differentiation to ATI cells. HIF-NOTCH and the fibrocyte growth factor receptor 2 signalings drive the expansion of KRT5+ epithelial progenitors (Nabhan et al., 2018).
The process of repairing the compromised tight junction in damaged alveoli is regulated by crosstalk between the extracellular matrix and the multiple alveolar cell types. Clinical evidence indicates the restorative role of injury-inducing immune cells in this process. Recruited fibroblast secretes epithelial growth factors depositing moderate quantities of collagen (Cong et al., 2017). Damages to mitochondrial complexes are destroyed, reported by immune cell functions, and replaced via mitochondrial transfer or biogenesis. The intercellular junctions are reassembled in a process regulated by multiple mechanisms. Mechanisms include signals from the basement membrane and beneficial effects from angiopoietin 1 (Wick et al., 2021). Once the tight junction reassembly is completed, edematous fluid in the alveoli is reabsorbed by diffusion through the water channels or by paracellular pathway transport into the interstitium. The osmotic gradient established by sodium uptake through the epithelial sodium channels and sodium transport through the Na+/K+-ATPase pumps drives the diffusion process.
In many patients, therapeutic interventions provide less-than-expected results, forcing care plans to adopt ventilation support and other management techniques. In a report on ARDS, Thompson et al. (2017) highlighted how the search for a consensus therapy plan for ARDS has been challenging despite huge investments in preclinical research. With the exemption of supporting care plans, including conservative fluid therapy and lung-protective ventilation, there have been difficulties adapting therapy success in animal models to human cases. Recently, a few research studies have investigated the difficulties in modeling therapy options in humans. These studies provide important clinical insight into biological markers, including plasma inflammatory markers and the inhibitory mechanism delaying alveoli repair in ARDS.
Researchers have identified many endogenous reparative mechanisms biologically inhibited during alveolar repair. As this mechanism becomes pronounced in some ARDS cases, the crosstalk between the extracellular matrix and the alveolar cell types is inhibited. Mitochondrial transfer, immune cell action, and the secretion of epithelial growth factors are inhibited, prompting symptomatic deterioration and a delay in tight junction reassembly. Depending on the etiology, the inhibitory mechanism implicated in alveolar repair varies. In ARDS cases precipitated by influenza, the virus infects KRT5+ progenitors and delays tight junction reassembly (Zhong et al., 2022). Hypercapnia, hypoxemia, influenza infections, and factors that downregulate the sodium channels or impair the Na+/K+-ATPase function also impair the mechanisms responsible for the removal of edematous fluid in the damaged alveolar (Gwoździńska et al., 2017). In hypercapnia, the alveolar epithelial cell proliferation is delayed. In animal models, keratinocyte growth factors reportedly increase the susceptibility of ATII cells to influenza virus infection and raise the mortality index (Nikolaidis et al., 2017).
The epidemiology of ARDS has long been studied to understand the pathophysiology in special populations. An early prospective analysis study focused on the incidence of ARDS in adults was conducted in the United States. The study design used validated screening protocols to identify patients (Gottlieb et al., 2022). The identifying criteria were defined as diagnosed non-Heart Failure ARDS using bilateral chest radiographic opacities and partial pressure of arterial oxygen to fraction of inspired oxygen ratio (PaO2/FiO2) greater than 300 mmHg. Using the data collated, the researchers reported an annual incidence of 190,000 ARDS cases in the United States with a mortality index of 38.5%. In a follow-up epidemiological study, the LUNG SAFE study provided data on ARDS epidemiology in a cross-section of 29,144 patients across 50 countries (Pham et al., 2017).
The prevalence of ARDS, as reported in the LUNG SAFE study, was pegged at 10% in intensive care patients and 23% in patients on ventilatory support (Pham et al., 2017). The study also reported a hospital mortality rate of 34.9% for patients with mild ARDS, 40% in patients with moderate ARDS, and 46.1% in patients with severe ARDS. There is limited research on how underlying comorbidities, including cancer and immunosuppression, influence mortality rates. A follow-up analysis of the study identified about 21% of the patients with clinical evidence of immunosuppression, and about 52% were non-immunocompromised patients (Cortegiani et al., 2018). Similar data from studies conducted between 2001 and 2008 suggest a decline in the incidence of ALI and ARDS in hospitalized adults (Song et al., 2021). The decline was partly related to the more widespread use of lung-protective ventilation, the adoption of multiple therapy plans, the use of blood products, and the reduction in hospital-acquired infections known to complicate the respiratory process (Toy et al., 2022).
Based on a recent ARDS Network trial published by the NIH HEART, Lung and Blood Institute (NHLBI), the 60-day mortality rate of ARDS has declined from 36% in 1997 to 26% in 2005. A decline of 22% in adult patients has also been recently published (Thomas et al., 2022).
Common conditions associated with an increased risk of ARDS include smoking, alcohol abuse, hypoalbuminemia, and air pollution (Reilly et al., 2019). For a reason that remains unclear, a low risk of ARDS has been identified in diabetic patients. Studies examining a possible link between the risk of ARDS and ethnicity have also been conducted. Available evidence shows a largely reported higher mortality rate in patients of Black and Hispanic descent. The rates are lower in White patients. On gender, male patients appear to have a higher ARDS-related mortality rate than women. However, the origins of these disparities remain largely under investigation.
Studies have also been designed to identify genetic factors that might be responsible for variations in the mortality rate of ARDS. Largely due to the phenotypic complexities of ARDS and the different risk factors involved, no specific loci associated with significant genetic variation for ARDS have been identified. However, candidate gene pathway analysis studies have identified important mechanisms linked with the mechanism of lung injuries that seem to influence the risk of ARDS. An example includes plasma angiopoietin 2, a protein marker and mediator of increased lung vascular permeability in a population of European ancestry (Reilly et al., 2018).
The Pediatric Acute Respiratory Distress Syndrome Incidence and Epidemiology (PARDIE) cross-sectional study of 145 international pediatric intensive care units is considered the reference ARDS epidemiological study in children. Based on the study results, the population-based incidence of ARDS in children aged 2 weeks to 17 years is estimated as 2.2-5.7 per 100,000. The condition is diagnosed in 2.3-3% of hospital pediatric intensive unit admission cases, with an estimated mortality rate of 17% - 33% (Khemani et al., 2019). Regarding gender variations in children, ARDS appears more frequently diagnosed in boys than in girls. As in adults, the etiologies are almost the same, with over 60% of pediatric ARDS linked with pneumonia. However, mortality risk seems lower in children, with higher rates only identified in those with indirect lung injury from sepsis and shock. In pediatric trauma cases, the incidence of ARDS is pegged at 0.5%, with a mortality rate of 18% (de Roulet et al., 2019). In pediatric cases involving intubation, the PARDIE study identified a significantly higher ARDS mortality rate compared with cases requiring no instance of intubation. A clinical history of cancer or hemopoietic stem cell transplantation also increases the mortality rate of pediatric ARDS to 43% compared to 11% in those without such a history.
The clinical presentation of ARDS is characterized by dyspnea and hypoxemia, which gradually worsen within 6 – 72 hours of clinical admission. In many cases, ventilation support of intensive care might be required within a few hours of presentation. In patients conscious enough to answer medical queries, complaints of mild dyspnea are often communicated as the respiratory condition worsens within 12 – 24 hours. Sometimes, mechanical ventilation might be urgently required to prevent hypoxia. Depending on the etiology, the precipitating cause or underlying condition may be identified, especially in cases of pneumonia and sepsis. In other cases, a detailed medical history might be needed to identify the instant case of respiratory distress. Generally, the physical presentation includes signs and symptoms associated with respiratory distress, including tachypnea and labored breathing. Systemic symptoms may also be evident depending on the severity of respiratory impairment. These include central or peripheral cyanosis, altered consciousness, and an altered mental status. Chest auscultations indicate rales, and the arterial oxygen saturation level remains low despite 100% oxygen supply.
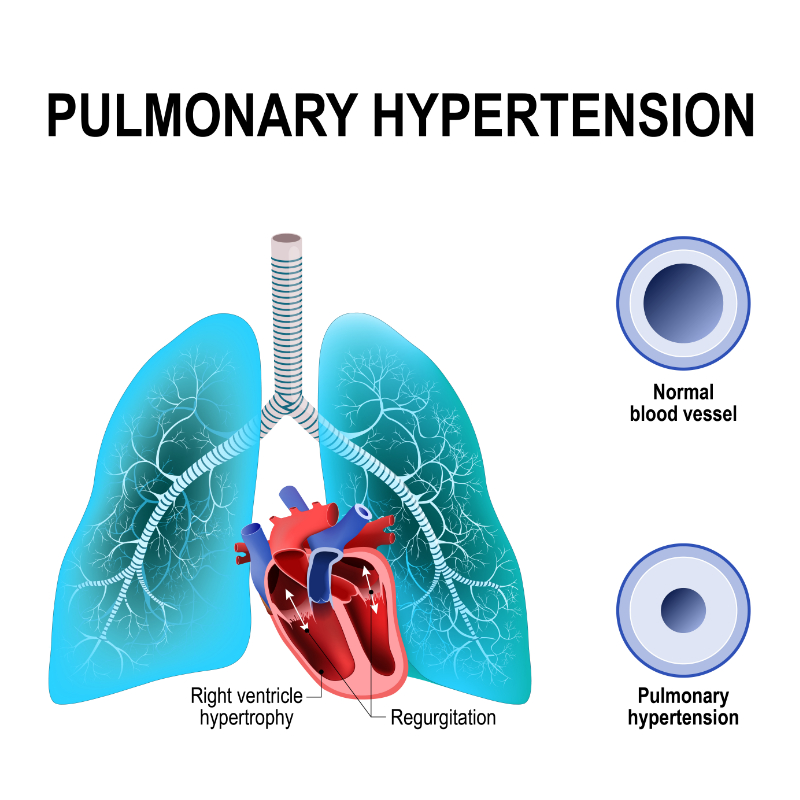
Pulmonary hypertension (PH) is another prominent characteristic feature of ARDS. ARDS involves a gradual increase in pulmonary vascular resistance, precipitating right ventricular dysfunction and ultimately causing right ventricular failure. Clinically, PH is defined as mean pulmonary arterial pressure (mPAP) greater than 20 mmHg measured by right heart catheterization (Simonneau et al., 2019). The most commonly studied underlying mechanisms of PH in ARDS include:
Pulmonary vasoconstriction
Vessel obliteration
Hypercapnia
An imbalance in vasoactive mediators
Micro thrombosis due to hypoxia (Price and Wort, 2017)
Pulmonary vasoconstriction, thromboembolism, and interstitial edema contribute to the development of PH. Damage to the lung causes neutrophils to release toxic mediators. These mediators, including reactive oxygen species, proteases, and pro-coagulation molecules, promote vasoconstriction and raise pulmonary vascular resistance. Price and Wort (2017) proposed that the pulmonary vascular injury noticed in PH is triggered by an increased inflammatory characteristic of ARDS secondary to ventilator-induced lung injury.
As a direct clinical consequence, PH may reduce systemic oxygen delivery, further impair tissue oxygen supply, and sharply raise the risk of organ dysfunction. The extent of PH also directly links with the severity of lung injury, prompting the need to resolve pulmonary hypertension in ARDS cases.
Image 5: Pulmonary Hypertension
With the recent adoption of mechanical ventilation in the management of ARDS, the incidence of PH in ARDS cases has reportedly declined. Mechanical ventilation lessens the intrathoracic airway pressure in ARDS and improves the low tidal volume and acute cor pulmonale in severe ARDS cases. However, despite advances in ARDS therapy, the prevalence of PH in ARDS remains high, with an epidemiological study reporting a prevalence of 46.6% in a 2016 survey. PH is often diagnosed as a complication of ARDS. However, no conclusive clinical evidence indicates a correlation between this condition and the severity of mortality of ARDS (Potus et al., 2020).
Since the presenting symptoms of ARDS can be nonspecific, there is a need to identify other medical conditions with almost the same symptoms. Collating patients' medical history and conducting a thorough physical examination investigating the physiological impairments of the respiratory and cardiovascular systems can help optimize diagnosis. Often, ARDS can be confused with congestive heart failure and pneumonia. In congestive heart failure, there is fluid overload, unlike in ARDS, where the symptomatology excludes signs of left atrial hypertension or volume overload. Also, other common signs of congestive heart failure are not evident in ARDS. These signs include edema, jugular venous distention, a salutary response to diuretics, third heart sound, and an elevated brain natriuretic peptide level.
Table 1: Factors distinguishing ARDS, CHF, and Pneumonia
Distinguishing Factor
ARDS
CHF
Pneumonia
Symptoms
Dyspnea
+
+
+
Hypoxia
+
+
+
Tachypnea
+
+
+
Pleuritic Chest Pain
+/-
-
+
Sputum Production
+/-
-
+
Signs
Rales
+
+
+
Fever
+/-
-
+
Edema
-
+
-
Jugular Venous Distention
-
+
-
Third Heart Sound
-
+
-
Studies
Hypoxemia
+
+
+
Bilateral Infiltrates
+
+/-
+/-
Pulmonary Wedge Pressure < 18 mm Hg
+
-
+
PaO2/FiO2 < 200
+
-
+
Localized Infiltrate
-
-
+
Elevated Brain Natriuretic Peptide
-
+
-
Cardiac Enlargement
-
+
-
Responses
Antibiotics
-
-
+
Diuretics
-
+
-
Oxygen
-
+
+
In pneumonia, the clinical findings are also slightly different. In uncomplicated pneumonia, signs of systemic and pulmonary inflammation may be evidenced by chills, fever, sputum production, pleuritic chest pain, fatigue, and localized or multifocal infiltrates. Also, in uncomplicated pneumonia, the accompanying case of hypoxia should respond to oxygen supplementation therapy. ARDS should be suspected in cases where oxygen supplementation does not improve accompanying hypoxia (Saguil et al., 2020). A definitive diagnosis is subject to other diagnostic criteria.
In uncontrolled sepsis or an advanced case of sepsis, pulmonary dysfunction may develop. There is often an associated risk of primary pulmonary complications, including pneumonia, exacerbation of COPD, aspiration, and pulmonary embolism (Jarczak et al., 2021). In cases of sepsis-induced multi-organ failure, the lung is a prominent organ system affected. A progression of sepsis-induced lung injury often develops into ARDS. Clinical surveys investigating the risk burden of ARDS complications in sepsis reported an incidence rate of 40% (Huppert et al., 2019). Available clinical evidence on this association has proposed different mechanisms for sepsis-induced ARDS. It is reportedly caused by multiple uncontrolled interactions between inflammatory cytokines and cellular mediators that impair the alveolar-capillary unit and disrupt the tight junction assembly.
Clinical studies on the mechanism of acute liver infection in sepsis-induced ARDS have identified multiple pathways. These include apoptosis, oxidative stress, endothelial barrier compromise, and inflammatory pathways. In sepsis, pathogenic factors induce inflammation by modulating signaling pathways that produce a huge volume of pro-inflammatory factors and mediators. Oxidative stress, on the other hand, increases the lung water content and damages the biological integrity of pulmonary endothelial barrier function. The damage to the alveolar capillaries in sepsis is induced by factors produced during oxidative stress, including nitrous oxide, PGE2, iNOS, COX-2, and SPD (Zhao et al., 2017). Endothelial cell apoptosis is a hallmark of sepsis that also destroys the tight junctions of the alveolar membrane. Protein factors also produced by apoptosis, such as caspase-3, can cause the rearrangement of cell junction proteins such as VE-cadherin.
Major trauma is identified as a risk factor for ARDS in inpatient admissions. Early clinical studies that identified this association also reported higher morbidity and raw mortality rates in trauma patients with ARDS complications than those without complications. A 2016 study examining the incidence, risk factors, and mortality from ARDS over a 2.5-year study period is considered the reference link between trauma and ARDS. The study reported an independent association between ARDS and emergent thoracotomy, blunt mechanism of traumatic injury, and fresh frozen transfusion. In another recent study, Kornblith et al. (2019) demonstrated that greater head and chest injury severity, crystalloid resuscitation, and a high volume of early platelet transfusion independently increase the risk of post-trauma ARDS (Kornblith et al., 2019). With the introduction of improved critical management and lower tidal volume ventilation, the overall mortality of trauma-induced ARDS has declined significantly in recent years.
In burn patients, ARDS may be precipitated via direct lung injury due to inhaled smoke and fumes. Inflammatory responses associated with burn and infectious complications may also increase the risk of post-burn ARDS (Mokra., 2020). An increase in capillary permeability in patients with extensive surface burns occurs beyond the lesion site and, in many cases, extends to the major organ close to the lesion site. Increased vascular permeability consequently leaks fluids into the interstitial space, triggering pulmonary edema and disruption to the alveolar membrane. The risk of ARDS in burn patients admitted to specialized intensive care units is high as this phenomenon can occur.
TRALI is an adverse event seen after transfusing blood products. Blood products include:
Whole blood
Fresh frozen plasma (FFP)
Platelets
Packed red blood cells
Granulocytes
Intravenous immune globulin
Autologous and allogenic stem cells
Cryoprecipitates (Suddock et al., 2022)
TRALI has an associated acute, noncardiogenic pulmonary edema linked with many pathologic mechanisms during or after transfusion. The FDA considers TRALI as the leading cause of death from transfusion-related medical care plans, with an incident of TRALI occurring in 1 out of every 5000 units of packed red blood cell transfused. Similar data estimates 1 event in 2000 plasma-containing component transfusion and 1 incident in every 400 units of whole-blood-derived platelet concentrates. The physical symptoms reported in these events establish a medical logic indicating ARDS as a complication of TRALI in many patients (Ariza-Prota et al., 2017).
In TRALI events, patients may experience fever, hypotension, tachycardia, and hypoxemia of SpO2 less than 90% on room air with a ratio of the partial pressure of oxygen to a fractional inspired oxygen concentration of less than 300 mmHg. The pathologic mechanism proposed for TRALI includes an immunologic mechanism characterized by the infusion of cytotoxic donor antibodies (HLA class I or II) directly against specific recipient leukocyte antigens. The opposing interaction causes complement activation, neutrophil (PMN)-mediated capillary leak, and disruption of the alveolar-capillary complex. The transfusion of anti-leukocyte antibodies or lipids from stored blood may activate sequestered PMNs, causing endothelial damage and capillary leak. Capillary leaks drive the absorption of fluids and protein into the alveolar space, resulting in alveolar edema and damage (Rebetz et al., 2018).
Drug-induced ARDS is a popular study focus in pulmonary medicine. The focus received a major boost with the introduction of clinical evidence demonstrating how some new molecular target drugs, including gefitinib and amiodarone, induce severe interstitial lung disease (You et al., 2022). Initial clinical case studies implicate methotrexate and certain herbal preparations as leading drug substances inducing ARDS in many patients. The most widely studied drug-related respiratory pathology results from pulmonary vascular insults, airway, and parenchymal conditions. Pulmonary vascular insults include endovascular infections, hemorrhage, and vasoconstrictive events. Airway conditions include bronchospasm and hemorrhage. Parenchymal conditions include aspiration-related events, pulmonary edema, hemorrhage, pneumothorax, and infectious and non-infectious pneumonitides. In ARDS, reported drug-induced alveolar damage is caused by an immunostimulatory mechanism and those precipitated by cytotoxic mechanisms (Anan et al., 2019).
Some narcotics, particularly methadone and morphine, have been implicated in developing pulmonary edema and increased capillary leak. Research studies have focused on long-term, high-dose use of cocaine, heroin, aspirin, cardiovascular drugs, epinephrine, and phenylephrine on pulmonary toxicity. Several neuropsychiatric drugs have also been implicated, including tricyclic antidepressants and phenothiazines. High-dose administration of tocolytic agents has increased the risk of noncardiogenic pulmonary edema. Idiosyncratic reactions, capillary leaks, and anaphylaxis are other pathologic mechanisms proposed in these cases. Some drugs damage pulmonary tissue by releasing inflammatory cytotoxic mediators and producing reactive oxygen species. Patients may present with dyspnea, tachypnea, and hypoxia associated with alveolar-filling radiographic findings.
Other popularly studied etiologies of ARDS include:
Infectious or aspiration pneumonia
Pancreatitis (clinical manifestations of acute pancreatitis)
Cardiothoracic surgery (postoperative pulmonary dysfunction)
In line with the 1994 AECC, the criteria for ARDS diagnosis include acute onset of hypoxia, noncardiogenic and bilateral infiltrates on chest radiographs, and the absence of left atrial hypertension. Hypoxemia, in this criterion, was quantified using the ratio of partial pressure of arterial oxygen and the fraction of inspired oxygen (Pao2/FiO2), with a Pao2/FiO2 < 200 mm Hg. With modification, the Berlin definition included:
Bilateral opacities observable on anteroposterior chest radiographs
A symptomatic period of at least one week following known clinical insult
Hypoxemia -defined by a Pao2/FiO2 < 300 mm Hg
A minimum PEEP ≥ 5 cm H2O, not fully explained by cardiac failure or fluid overload
A CT scan may be required to correctly identify infiltrates as pulmonic.
A comprehensive review of physical presentation, medical history, radiographic imaging, and laboratory tests is required to evaluate the risk burden and possible etiology and make an informed diagnosis in ARDS management. If pulmonary infections are suspected, the typical diagnostic approach includes microscopic examination, respiratory specimen culture, and blood cultures. The medical history may identify a class of likely causative organisms or pathogens in community-acquired or hospital-acquired ARDS etiologies. Depending on geographic distribution and host immunity, the most commonly isolated organism in sepsis- and infection-induced ARDS include:
The Middle East respiratory syndrome coronavirus (MERS-CoV)
Plain chest radiography can help support diagnosis, monitor clinical progression, and evaluate the risk of clinical complications. Depending on the stages of ARDS, the plain chest radiography result may present differing radiographic outlines. In the first 24 hours, there is usually a latent radiographic period with a frequently normal radiography outline. In the following 24-72 hours, there is rapid deterioration as pulmonary edema and the disruption of the alveolar-capillary complex progresses. Air spaces and interstitial opacities on chest radiographs are predominantly symmetrical and bilateral (Huang et al., 2022). The presence of peripheral alveolar opacities supports the diagnosis of ARDS, as well as the lack of temporal change and the absence of cardiomegaly, septal lines, and pleural fluid. During the proliferative stage (8-14 days), the radiographic outline usually stabilizes for a variable period. Diffuse coarse reticular opacities may develop but also may resolve. The chest radiograph may be completely normal, or there may be widespread coarse reticular opacities.
During the early phase of ARDS, the CT tends to show:
An opacification that demonstrates an anterior-posterior density gradient within the lung
Dense consolidation in the most dependent regions, merging into a background of widespread ground-glass attenuation
Normal or hyperexpanded lung in the non-dependent regions
The CT findings likely represent edema and protein within the interstitium and alveoli in acute ARDS. Another important observed feature in acute ARDS is bronchial dilatation within areas of ground-glass opacification. Although this is considered an indicator of early fibrosis, this is not conclusive proof of fibrosis, as the reversal of bronchial dilation in the later stages of ARDS is well known. CT appearances become variable after the acute phase of ARDS, and complete resolution of abnormalities may occur. A coarse reticular pattern and ground-glass opacification in the lung's anterior (non-dependent) part may be seen. Pulmonary cysts of varying sizes and bullae are also features of the later stages of ARDS and probably develop due to prolonged ventilation (Regli et al., 2021).
Beyond tracing disease progression, CT can help identify various ventilation-association complications and foci of infection that may be difficult to evaluate on the supine, anteroposterior, intensive care unit radiography. In addition, the sequelae of mechanical ventilation may also be easily detected on CT. Distinct CT results may detect pneumonia, abscesses, pneumothorax, pulmonary interstitial emphysema, and pneumomediastinum.
In 2018, the guidelines for the management of ARDS were drafted by a multidisciplinary writing group constituted by the Joint Standards Committee of the Faculty of Intensive Care Medicine and Intensive Care Services. Since the first draft, this guideline has been extensively reviewed by stakeholders of the medical community and independent reviewers. The guideline focused on ten topics based on a committee member's existing recommendation regarding ARDS management. These topics include:
Corticosteroids
ECMO
ECCO2R
Fluid strategy
High-frequency oscillation ventilation (HFOV)
Inhaled vasodilators (iVasoD)
Lung protective ventilation: tidal volume (Vt)
Neuromuscular blocking agent (NMBA)
PEEP
Prone Positioning
Using the PICO (Population, Intervention, Comparison, Outcome) formulation, each topic was developed into full protocols, using systemic reviews and meta-analyses of available clinical evidence on ARDS. The management of ARDS in its entirety is focused on respiratory support, fluid management, nutritional supplementation, and post-therapy rehabilitation.
Primarily, mechanical ventilation's endpoint in managing ARDS is maintaining optimal oxygenation and eliminating carbon dioxide from the respiratory flow. In a 2000 randomized phase III trial sponsored by the US National Heart Lung and Blood Institute ARDS Network, researchers reported an improved clinical outcome with a tidal volume of 6 ml per kg of PBW, compared with the common higher tidal volume of 12 ml per kg of PBW. The tidal volume improved the survival rate, shortened mechanical ventilation duration, accelerated extrapulmonary organ failure recovery, and decreased systemic inflammation's clinical burden (Frat et al., 2022).
A tidal volume of around 6 mL/kg of predicted body weight PBW should be used as the first approach in patients with recognized ARDS, in the absence of severe metabolic acidosis, including those with mild ARDS.
The experts suggest a similar approach for all patients on invasive mechanical ventilation and under sedation in ICU, given the high rate of failure to recognize ARDS and the importance of rapidly implementing pulmonary protection.
Once the tidal volume is set to around 6 mL/kg PBW, plateau pressure should be monitored continuously and not exceed 30 cmH2O.
The experts suggest that tidal volume should not be increased when the plateau pressure is well below 30 cmH2O, except in marked, persistent hypercapnia, despite reduced instrumental dead space and increased respiratory rate.
By recommendation, lower tidal volumes are needed in ARDS management to prevent regional overdistension. However, scaling tidal volume to PBW targets estimated healthy lung size, although aerated baby lung volume can differ substantially between patients. Targeting airway driving pressure (plateau pressure minus PEEP) is one strategy for tailoring tidal volume to patient-specific mechanics that has garnered considerable attention. For patients with mild lung injury at a lower risk of biophysical injury, the benefit of lowering tidal volume should be weighed against the risks of more aggressive sedation or the use of paralytics if needed to achieve the intended tidal volumes.
PEEP is an essential component of managing ARDS, and the experts suggest using a value above 5 cmH2O in all patients presenting with ARDS.
High PEEP should probably be used in patients with moderate or severe ARDS but not in patients with mild ARDS.
The experts suggest reserving high PEEP for patients in whom it improves oxygenation without marked deterioration. PEEP settings should be individualized.
PEEP is considered an integral component of protective ventilation. Adopting PEEP recommendations in ARDS management aims to ensure optimized oxygenation and maintain alveolar recruitment. In ARDS patients, the ideal PEEP sufficiently operates the cyclic opening and collapse of distal airspaces during tidal ventilation and is low enough to avoid tidal overdistension. It is important to identify distal airspaces of the lung that may be collapsed or edematous and can be inflated with higher levels of PEEP to improve gas exchange. However, caution is advised with these methods. Cavalcanti et al. (2017) reported the worst outcomes with a strategy of aggressive recruitment maneuvers and very high PEEP in patients with moderate or severe ARDS, receiving 6 ml per kg PBW tidal volume.
Prone positioning should be used in ARDS patients with PaO2/FIO2 ratio < 150 mmHg to reduce mortality. Sessions of at least 16 consecutive hours should be performed.
Prone positioning has been studied in randomized control trials and meta-analyses focusing on understanding any potential difference in mortality between prone and supine position groups (Munshi et al., 2017). Prone positioning decreases lung aeration by modifying the distribution of transpulmonary pressure. A decrease in pressure reduces the risk of mechanical lung injury and improves gas exchange. The multicenter Proning Severe ARDS Patients (PROSEVA) trial is a reference study examining the efficacy of prone positioning as respiratory support in managing ARDS. The study enrolled patients with moderate or severe ARDS, with prone positioning prescribed for at least 16 hours daily. The study results provided conclusive evidence demonstrating how prone positioning improved survival and shortened the duration of mechanical ventilation compared to other positioning techniques (Fan et al., 2017).
To reduce mortality, a neuromuscular blocking agent should probably be considered in ARDS patients with a PaO2/FiO2 ratio < 150 mmHg. The neuromuscular blocking agent should be administered by continuous infusion early (within 48 h after the start of ARDS), for no more than 48 h, with at least daily evaluation.
The focus of neuromuscular blocking agents as respiratory support in ARDS management is on testing the effect of these agents on deep sedation at the acute phase. In patients with acute ARDS, elevated transpulmonary pressures may increase the risk of lung injury. In addition, ventilated patients may exhibit strong respiratory effort when high doses of sedatives are administered. The synergistic effect of strong respiratory effort and elevated transpulmonary pressures may result in increased mechanical lung injuries. Paralysis induced by neuromuscular blocking agents (NBA) may decrease this strong respiratory effort and reduce the risk of mechanical injuries. Clinical studies testing this hypothesis using cisatracurium as an NBA have reported improved adjusted survival and more ventilator-free days.
Rescue therapies are instituted in patients experiencing continuous clinical deterioration despite an optimized management plan. These therapies complement the efficacy of respiratory support therapies in ARDS patients.
Veno-venous extracorporeal membrane oxygenation (ECMO) should be considered in cases of severe ARDS with PaO2/FiO2 < 80 mmHg. ECMO should also be considered when mechanical ventilation becomes dangerous because of the increase in plateau pressure and despite optimization of ARDS management, including high PEEP, neuromuscular blocking agents, and prone positioning. The decision to use ECMO should be evaluated early through contact with an expert center.
With ECMO, blood is circulated outside the body for oxygenation on a gas-permeable membrane. It is considered a first-choice rescue therapy in patients with very severe ARDS, in whom sufficient correction of gas exchange is inconsistent with lung-protective ventilation. ECMO is recommended in patients with refractory hypoxemia due to pulmonary failure.
Combes et al. (2018) reported an 11% absolute reduction in 60-day mortality with ECMO with ventilator settings targeting very low tidal volumes to achieve a plateau airway pressure of less than 24 cmH2O, compared with the now conventional strategy of 6 ml per kg PBW tidal volumes and plateau airway pressures up to 30 cmH2O (Combes et al., 2018). The best potential candidates for ECMO are patients with severe ARDS within the first week of mechanical ventilation and without multiple organ failures. It is recommended that ECMO should only be performed in centers with adequate extracorporeal support and the proper clinical requirements for managing severe ARDS (Mi et al., 2018). Extracorporeal carbon dioxide removal is another rescue therapy that can be considered in severe ARDS.
Nutritional support might be instituted as a rehabilitation care plan in ARDS management. A large percentage of ARDS patients might experience postoperative difficulty in eating and exhibiting normal swallowing reflexes. Enteral and parenteral feeding may be instituted depending on the state of the gastrointestinal tract. Meal plans with high-fat and low-carbohydrate are recommended due to their vasodilatory and anti-inflammatory effects.
Tracheostomy and percutaneous endoscopic gastrostomy (PEG) are required in the recovery phase, as many ARDS patients may require feedings and the removal of secretions. Rehabilitation also facilitates oxygen weaning from the ventilator making supplemental oxygen discontinuation easier and more comfortable. Tracheostomy is done at week 2 or 3, followed by percutaneous feeding.
Moderated activity in rehabilitation care in ARDS reduces the risk of venous thrombosis, stress-induced ulcers, and bedsores in bed-bound patients. Activities can be done with frequent position changes, timed ambulation, and decreased sedation time.
Select one of the following methods to complete this course.
Take TestPass an exam testing your knowledge of the course material.
CEUFast, Inc. is committed to furthering diversity, equity, and inclusion (DEI). While reflecting on this course content, CEUFast, Inc. would like you to consider your individual perspective and question your own biases. Remember, implicit bias is a form of bias that impacts our practice as healthcare professionals. Implicit bias occurs when we have automatic prejudices, judgments, and/or a general attitude towards a person or a group of people based on associated stereotypes we have formed over time. These automatic thoughts occur without our conscious knowledge and without our intentional desire to discriminate. The concern with implicit bias is that this can impact our actions and decisions with our workplace leadership, colleagues, and even our patients. While it is our universal goal to treat everyone equally, our implicit biases can influence our interactions, assessments, communication, prioritization, and decision-making concerning patients, which can ultimately adversely impact health outcomes. It is important to keep this in mind in order to intentionally work to self-identify our own risk areas where our implicit biases might influence our behaviors. Together, we can cease perpetuating stereotypes and remind each other to remain mindful to help avoid reacting according to biases that are contrary to our conscious beliefs and values.
Abdulnour, R. E., Gunderson, T., Barkas, I., Timmons, J. Y., Barnig, C., Gong, M., Kor, D. J., Gajic, O., Talmor, D., Carter, R. E., & Levy, B. D. (2018). Early Intravascular Events Are Associated with Development of Acute Respiratory Distress Syndrome. A Substudy of the LIPS-A Clinical Trial. American journal of respiratory and critical care medicine, 197(12), 1575–1585. Visit Source.
Anan, K., Ichikado, K., Ishihara, T., Shintani, A., Kawamura, K., Suga, M., & Sakagami, T. (2019). A Scoring System with High-Resolution Computed Tomography to Predict Drug-Associated Acute Respiratory Distress Syndrome: Development and Internal Validation. Scientific reports, 9(1), 8601. Visit Source.
Ariza-Prota, M. A., Pando-Sandoval, A., & Budiño, T. (2017). Transfusion related acute lung injury: An underdiagnosed and dangerous entity. Revista portuguesa de pneumologia, 23(3), 160–161. Visit Source.
Beretta, E., Romanò, F., Sancini, G., Grotberg, J. B., Nieman, G. F., & Miserocchi, G. (2021). Pulmonary Interstitial Matrix and Lung Fluid Balance From Normal to the Acutely Injured Lung. Frontiers in physiology, 12, 781874. Visit Source.
Cardinal-Fernández, P., Lorente, J. A., Ballén-Barragán, A., & Matute-Bello, G. (2017). Acute Respiratory Distress Syndrome and Diffuse Alveolar Damage. New Insights on a Complex Relationship. Annals of the American Thoracic Society, 14(6), 844–850. Visit Source.
Chen, X., Cheng, K., Sun, X., Zhang, Y., Cao, Z., Li, J., Bai, J., Lu, H., Gu, S., Zhang, L., Xu, J., Jiang, P., & Liang, S. (2021). Comparison of traditional methods and high-throughput genetic sequencing in the detection of pathogens in pulmonary infectious diseases. Annals of translational medicine, 9(8), 702. Visit Source.
Combes, A., Hajage, D., Capellier, G., Demoule, A., Lavoué, S., Guervilly, C., Da Silva, D., Zafrani, L., Tirot, P., Veber, B., Maury, E., Levy, B., Cohen, Y., Richard, C., Kalfon, P., Bouadma, L., Mehdaoui, H., Beduneau, G., Lebreton, G., Brochard, L., … EOLIA Trial Group, REVA, and ECMONet (2018). Extracorporeal Membrane Oxygenation for Severe Acute Respiratory Distress Syndrome. The New England journal of medicine, 378(21), 1965–1975. Visit Source.
Cong, X., Hubmayr, R. D., Li, C., & Zhao, X. (2017). Plasma membrane wounding and repair in pulmonary diseases. American journal of physiology. Lung cellular and molecular physiology, 312(3), L371–L391. Visit Source.
Cortegiani, A., Madotto, F., Gregoretti, C., Bellani, G., Laffey, J. G., Pham, T., Van Haren, F., Giarratano, A., Antonelli, M., Pesenti, A., Grasselli, G., & LUNG SAFE Investigators and the ESICM Trials Group (2018). Immunocompromised patients with acute respiratory distress syndrome: secondary analysis of the LUNG SAFE database. Critical care (London, England), 22(1), 157. Visit Source.
Davidson, K. R., Ha, D. M., Schwarz, M. I., & Chan, E. D. (2020). Bronchoalveolar lavage as a diagnostic procedure: a review of known cellular and molecular findings in various lung diseases. Journal of thoracic disease, 12(9), 4991–5019. Visit Source.
de Roulet, A., Burke, R. V., Lim, J., Papillon, S., Bliss, D. W., Ford, H. R., Upperman, J. S., Inaba, K., & Jensen, A. R. (2019). Pediatric trauma-associated acute respiratory distress syndrome: Incidence, risk factors, and outcomes. Journal of pediatric surgery, 54(7), 1405–1410. Visit Source.
Doig, C., Cooke, R., Chua, C., & Leung, T. (2022). Acute respiratory distress syndrome precipitated by granulocyte colony-stimulating factor in undiagnosed Pneumocystis jirovecii pneumonia. BMJ case reports, 15(2), e242316. Visit Source.
Esquivel-Ruiz, S., González-Rodríguez, P., Lorente, J. A., Pérez-Vizcaíno, F., Herrero, R., & Moreno, L. (2021). Extracellular Vesicles and Alveolar Epithelial-Capillary Barrier Disruption in Acute Respiratory Distress Syndrome: Pathophysiological Role and Therapeutic Potential. Frontiers in physiology, 12, 752287. Visit Source.
Fan, E., Del Sorbo, L., Goligher, E. C., Hodgson, C. L., Munshi, L., Walkey, A. J., Adhikari, N., Amato, M., Branson, R., Brower, R. G., Ferguson, N. D., Gajic, O., Gattinoni, L., Hess, D., Mancebo, J., Meade, M. O., McAuley, D. F., Pesenti, A., Ranieri, V. M., Rubenfeld, G. D., … American Thoracic Society, European Society of Intensive Care Medicine, and Society of Critical Care Medicine (2017). An Official American Thoracic Society/European Society of Intensive Care Medicine/Society of Critical Care Medicine Clinical Practice Guideline: Mechanical Ventilation in Adult Patients with Acute Respiratory Distress Syndrome. American journal of respiratory and critical care medicine, 195(9), 1253–1263. Visit Source.
Frat, J. P., Le Pape, S., Coudroy, R., & Thille, A. W. (2022). Noninvasive Oxygenation in Patients with Acute Respiratory Failure: Current Perspectives. International journal of general medicine, 15, 3121–3132. Visit Source.
Gottlieb, M., Chesis, M., & Long, B. (2022, May). What is the Impact of Low Tidal Volume Ventilation for Emergency Department Patients? Annals of Emergency Medicine. Visit Source.
Gwoździńska, P., Buchbinder, B. A., Mayer, K., Herold, S., Morty, R. E., Seeger, W., & Vadász, I. (2017). Hypercapnia Impairs ENaC Cell Surface Stability by Promoting Phosphorylation, Polyubiquitination and Endocytosis of β-ENaC in a Human Alveolar Epithelial Cell Line. Frontiers in immunology, 8, 591. Visit Source.
Huang, S., Wang, Y. C., & Ju, S. (2022). Advances in medical imaging to evaluate acute respiratory distress syndrome. Chinese journal of academic radiology, 5(1), 1–9. Visit Source.
Huppert, L. A., Matthay, M. A., & Ware, L. B. (2019). Pathogenesis of Acute Respiratory Distress Syndrome. Seminars in respiratory and critical care medicine, 40(1), 31–39. Visit Source.
Hussain, M., Khurram Syed, S., Fatima, M., Shaukat, S., Saadullah, M., Alqahtani, A. M., Alqahtani, T., Bin Emran, T., Alamri, A. H., Barkat, M. Q., & Wu, X. (2021). Acute Respiratory Distress Syndrome and COVID-19: A Literature Review. Journal of inflammation research, 14, 7225–7242. Visit Source.
Jarczak, D., Kluge, S., & Nierhaus, A. (2021). Sepsis-Pathophysiology and Therapeutic Concepts. Frontiers in medicine, 8, 628302. Visit Source.
Jones, J. H., & Minshall, R. D. (2022). Endothelial Transcytosis in Acute Lung Injury: Emerging Mechanisms and Therapeutic Approaches. Frontiers in physiology, 13, 828093. Visit Source.
Kaku, S., Nguyen, C. D., Htet, N. N., Tutera, D., Barr, J., Paintal, H. S., & Kuschner, W. G. (2020). Acute Respiratory Distress Syndrome: Etiology, Pathogenesis, and Summary on Management. Journal of intensive care medicine, 35(8), 723–737. Visit Source.
Kao, K. C., Chang, C. H., Hung, C. Y., Chiu, L. C., Huang, C. C., & Hu, H. C. (2017). Survival predictor in patients with acute respiratory distress syndrome and diffuse alveolar damage undergoing open lung biopsy. PloS one, 12(7), e0180018. Visit Source.
Kersten, A., & Cornelissen, C. (2020). Acute respiratory distress syndrome. Der Pneumologe, 17(4), 238–248. Visit Source.
Khemani, R. G., Smith, L., Lopez-Fernandez, Y. M., Kwok, J., Morzov, R., Klein, M. J., Yehya, N., Willson, D., Kneyber, M., Lillie, J., Fernandez, A., Newth, C., Jouvet, P., Thomas, N. J., Pediatric Acute Respiratory Distress syndrome Incidence and Epidemiology (PARDIE) Investigators, & Pediatric Acute Lung Injury and Sepsis Investigators (PALISI) Network (2019). Paediatric acute respiratory distress syndrome incidence and epidemiology (PARDIE): an international, observational study. The Lancet. Respiratory medicine, 7(2), 115–128.Visit Source.
Kornblith, L. Z., Robles, A. J., Conroy, A. S., Redick, B. J., Howard, B. M., Hendrickson, C. M., Moore, S., Nelson, M. F., Moazed, F., Callcut, R. A., Calfee, C. S., & Cohen, M. J. (2019). Predictors of postinjury acute respiratory distress syndrome: Lung injury persists in the era of hemostatic resuscitation. The journal of trauma and acute care surgery, 87(2), 371–378. Visit Source.
Lefrançais, E., Mallavia, B., Zhuo, H., Calfee, C. S., & Looney, M. R. (2018). Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI insight, 3(3), e98178. Visit Source.
Liberti, D. C., Kremp, M. M., Liberti, W. A., 3rd, Penkala, I. J., Li, S., Zhou, S., & Morrisey, E. E. (2021). Alveolar epithelial cell fate is maintained in a spatially restricted manner to promote lung regeneration after acute injury. Cell reports, 35(6), 109092. Visit Source.
Lupu, L., Palmer, A., & Huber-Lang, M. (2020). Inflammation, Thrombosis, and Destruction: The Three-Headed Cerberus of Trauma- and SARS-CoV-2-Induced ARDS. Frontiers in immunology, 11, 584514. Visit Source.
Matthay, M. A., Thompson, B. T., & Ware, L. B. (2021). The Berlin definition of acute respiratory distress syndrome: should patients receiving high-flow nasal oxygen be included? The Lancet Respiratory Medicine, 9(8), 933–936. Visit Source.
Meyer, N. J., Gattinoni, L., & Calfee, C. S. (2021). Acute respiratory distress syndrome. Lancet (London, England), 398(10300), 622–637. Visit Source.
Mi, M. Y., Matthay, M. A., & Morris, A. H. (2018). Extracorporeal Membrane Oxygenation for Severe Acute Respiratory Distress Syndrome. The New England journal of medicine, 379(9), 884–887. Visit Source.
Mokrá D. (2020). Acute lung injury - from pathophysiology to treatment. Physiological research, 69(Suppl 3), S353–S366. Visit Source.
Munshi, L., Del Sorbo, L., Adhikari, N., Hodgson, C. L., Wunsch, H., Meade, M. O., Uleryk, E., Mancebo, J., Pesenti, A., Ranieri, V. M., & Fan, E. (2017). Prone Position for Acute Respiratory Distress Syndrome. A Systematic Review and Meta-Analysis. Annals of the American Thoracic Society, 14(Supplement_4), S280–S288. Visit Source.
Nabhan, A. N., Brownfield, D. G., Harbury, P. B., Krasnow, M. A., & Desai, T. J. (2018). Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science (New York, N.Y.), 359(6380), 1118–1123. Visit Source.
Nikolaidis, N. M., Noel, J. G., Pitstick, L. B., Gardner, J. C., Uehara, Y., Wu, H., Saito, A., Lewnard, K. E., Liu, H., White, M. R., Hartshorn, K. L., & McCormack, F. X. (2017). Mitogenic stimulation accelerates influenza-induced mortality by increasing susceptibility of alveolar type II cells to infection. Proceedings of the National Academy of Sciences of the United States of America, 114(32), E6613–E6622. Visit Source.
Ortiz, G., Garay, M., Capelozzi, V., & Cardinal-Fernández, P. (2019). Airway Pathological Alterations Selectively Associated With Acute Respiratory Distress Syndrome and Diffuse Alveolar Damage - Narrative Review. Archivos de bronconeumologia, 55(1), 31–37. Visit Source.
Patel, S. R., Harris, R. S., & Malhotra, A. (2002). Pulmonary dead space and survival. The New England journal of medicine, 347(11), 850–852.
Pham, T., & Rubenfeld, G. D. (2017). Fifty Years of Research in ARDS. The Epidemiology of Acute Respiratory Distress Syndrome. A 50th Birthday Review. American journal of respiratory and critical care medicine, 195(7), 860–870. Visit Source.
Potus, F., Mai, V., Lebret, M., Malenfant, S., Breton-Gagnon, E., Lajoie, A. C., Boucherat, O., Bonnet, S., & Provencher, S. (2020). Novel insights on the pulmonary vascular consequences of COVID-19. American journal of physiology. Lung cellular and molecular physiology, 319(2), L277–L288. Visit Source.
Price, L. C., & Wort, S. J. (2017). Pulmonary hypertension in ARDS: inflammation matters! Thorax, 72(5), 396–397. Visit Source.
Rebetz, J., Semple, J. W., & Kapur, R. (2018). The Pathogenic Involvement of Neutrophils in Acute Respiratory Distress Syndrome and Transfusion-Related Acute Lung Injury. Transfusion medicine and hemotherapy: offizielles Organ der Deutschen Gesellschaft fur Transfusionsmedizin und Immunhamatologie, 45(5), 290–298. Visit Source.
Regli, A., Ahmadi-Noorbakhsh, S., Musk, G. C., Reese, D. J., Herrmann, P., Firth, M. J., & Pillow, J. J. (2021). Computed tomographic assessment of lung aeration at different positive end-expiratory pressures in a porcine model of intra-abdominal hypertension and lung injury. Intensive care medicine experimental, 9(1), 52. Visit Source.
Reilly, J. P., Wang, F., Jones, T. K., Palakshappa, J. A., Anderson, B. J., Shashaty, M., Dunn, T. G., Johansson, E. D., Riley, T. R., Lim, B., Abbott, J., Ittner, C., Cantu, E., Lin, X., Mikacenic, C., Wurfel, M. M., Christiani, D. C., Calfee, C. S., Matthay, M. A., Christie, J. D., … Meyer, N. J. (2018). Plasma angiopoietin-2 as a potential causal marker in sepsis-associated ARDS development: evidence from Mendelian randomization and mediation analysis. Intensive care medicine, 44(11), 1849–1858. Visit Source.
Reilly, J. P., Zhao, Z., Shashaty, M., Koyama, T., Christie, J. D., Lanken, P. N., Wang, C., Balmes, J. R., Matthay, M. A., Calfee, C. S., & Ware, L. B. (2019). Low to Moderate Air Pollutant Exposure and Acute Respiratory Distress Syndrome after Severe Trauma. American journal of respiratory and critical care medicine, 199(1), 62–70. Visit Source.
Saguil, A., & Fargo, M. V. (2020). Acute Respiratory Distress Syndrome: Diagnosis and Management. American family physician, 101(12), 730–738.
Simonneau, G., Montani, D., Celermajer, D. S., Denton, C. P., Gatzoulis, M. A., Krowka, M., Williams, P. G., & Souza, R. (2019). Haemodynamic definitions and updated clinical classification of pulmonary hypertension. The European respiratory journal, 53(1), 1801913. Visit Source.
Song, X., Weister, T. J., Dong, Y., Kashani, K. B., & Kashyap, R. (2021). Derivation and Validation of an Automated Search Strategy to Retrospectively Identify Acute Respiratory Distress Patients Per Berlin Definition. Frontiers in medicine, 8, 614380. Visit Source.
Suddock J.T., Crookston K. P. Transfusion Reactions. (2022). In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Visit Source.
Thille, A. W., Peñuelas, O., Lorente, J. A., Fernández-Segoviano, P., Rodriguez, J. M., Aramburu, J. A., Panizo, J., Esteban, A., & Frutos-Vivar, F. (2017). Predictors of diffuse alveolar damage in patients with acute respiratory distress syndrome: a retrospective analysis of clinical autopsies. Critical care (London, England), 21(1), 254. Visit Source.
Thomas, R., Turaka, V. P., Peter, J. V., Christopher, D. J., Balamugesh, T., Mahasampath, G., Mathuram, A. J., Sadiq, M., Ramya, I., George, T., Chandireseharan, V., George, T., & Sudarsanam, T. D. (2022). Good survival rate, moderate overall and good respirator quality of life, near normal pulmonary functions, and good return to work despite catastrophic economic costs 6 months following recovery from Acute Respiratory Distress Syndrome. Lung India: official organ of Indian Chest Society, 39(2), 169–173. Visit Source.
Thompson, B. T., Chambers, R. C., & Liu, K. D. (2017). Acute Respiratory Distress Syndrome. The New England journal of medicine, 377(6), 562–572. Visit Source.
Toy, P., Looney, M. R., Popovsky, M., Palfi, M., Berlin, G., Chapman, C. E., Bolton-Maggs, P., & Matthay, M. A. (2022). Transfusion-related Acute Lung Injury: 36 Years of Progress (1985-2021). Annals of the American Thoracic Society, 19(5), 705–712. Visit Source.
Wick, K. D., Mcauley, D. F., Levitt, J. E., Beitler, J. R., Annane, D., Riviello, E. D., Calfee, C. S., & Matthay, M. A.. (2021). Promises and challenges of personalized medicine to guide ARDS therapy. Critical Care, 25(1). Visit Source.
Writing Group for the Alveolar Recruitment for Acute Respiratory Distress Syndrome Trial (ART) Investigators, Cavalcanti, A. B., Suzumura, É. A., Laranjeira, L. N., Paisani, D. M., Damiani, L. P., Guimarães, H. P., Romano, E. R., Regenga, M. M., Taniguchi, L., Teixeira, C., Pinheiro de Oliveira, R., Machado, F. R., Diaz-Quijano, F. A., Filho, M., Maia, I. S., Caser, E. B., Filho, W. O., Borges, M. C., Martins, P. A., … Ribeiro de Carvalho, C. R. (2017). Effect of Lung Recruitment and Titrated Positive End-Expiratory Pressure (PEEP) vs Low PEEP on Mortality in Patients With Acute Respiratory Distress Syndrome: A Randomized Clinical Trial. JAMA, 318(14), 1335–1345. Visit Source.
Wu, Y., Yang, X., Ju, Y., & Zhao, F. (2022). Fraxinol attenuates LPS-induced acute lung injury by equilibrating ACE-Ang II-AT1R and ACE2-Ang (1-7)-Mas and inhibiting NLRP3. Pharmaceutical biology, 60(1), 979–989. Visit Source.
Xie, Y., Yu, Y., Zhao, L., Ning, P., Luo, Q., Zhang, Y., Yin, L., Zheng, Y., & Gao, Z. (2021). Specific Cytokine Profiles Predict the Severity of Influenza A Pneumonia: A Prospectively Multicenter Pilot Study. BioMed research international, 2021, 9533044. Visit Source.
You, H. S., Yoon, J. H., Cho, S. B., Choi, Y. D., Kim, Y. H., Choi, W., Kang, H. C., & Choi, S. K. (2022). Amiodarone-Induced Multi-Systemic Toxicity Involving the Liver, Lungs, Thyroid, and Eyes: A Case Report. Frontiers in cardiovascular medicine, 9, 839441. Visit Source.
Zhao, B., Gao, W., Gao, X., Leng, Y., Liu, M., Hou, J., & Wu, Y. (2017). Sulforaphane attenuates acute lung injury by inhibiting oxidative stress via Nrf2/HO-1 pathway in a rat sepsis model. International journal of clinical and experimental pathology, 10(8), 9021–9028.
Zhong, Q., Liu, Y., Correa, M. R., Marconett, C. N., Minoo, P., Li, C., Ann, D. K., Zhou, B., & Borok, Z. (2022). FOXO1 Couples KGF and PI-3K/AKT Signaling to NKX2.1-Regulated Differentiation of Alveolar Epithelial Cells. Cells, 11(7), 1122. Visit Source.
Listen from your pocket or in your car, Apple CarPlay and Android Auto compatible.
Sync Between Devices
Start on your desktop, pickup on your mobile. Never lose your place.
Reinforce Your Learning
Switch between reading and listening to your course or read while you listen!
Enhance Your Experience
Interactive course outline, robust play controls, multiple voices and different playback speed options.
Listen Anywhere
Listen from your pocket or in your car, Apple CarPlay and Android Auto compatible.
Subscribe for Access
Sync Between Devices
Start on your desktop, pickup on your mobile. Never lose your place.
Reinforce Your Learning
Switch between reading and listening to your course or read while you listen!
Enhance Your Experience
Interactive course outline, robust play controls, multiple voices and different playback speed options.
×
Away for now!
We're sorry! All our support agents are unavailable to chat at the moment.
Need Immediate Assistance? Please visit our FAQ section which provides answers to many common inquiries.
Get in Touch Directly via Email If your query is urgent or you'd prefer to reach us directly, we invite you to submit a support ticket through our Contact Us page. Our dedicated support team will review your inquiry and get back to you as soon as possible.
Support Hours Our chat support is typically available Monday to Friday, 9:00 AM - 6:00 PM EST. For support outside of these hours, please use our email contact
Are you sure?
You should NOT select "Remember me for 30 days" if you are on a public or shared computer.