For many, a diagnosis of Marfan syndrome is the missing puzzle piece that explains a lifetime of unrelated symptoms: a racing heart, unusual height, or sudden vision changes. It is more than a medical label; it is a genetic reality that shapes the daily lives of thousands of families, influencing everything from career choices to family planning.
February serves as National Marfan Syndrome Awareness Month, a crucial time to bring attention to this serious genetic condition. Marfan syndrome is an inherited connective tissue disorder affecting approximately 1 in 5,000 individuals worldwide. This article reviews the physical features, diagnostic considerations, and current management strategies that have improved the prognosis for those living with the condition.
Who Is Affected by Marfan Syndrome?
This syndrome affects individuals regardless of sex, age, race, or ethnicity. The condition occurs worldwide with equal frequency across all populations and geographic regions.
Autosomal dominant inheritance pattern:
A child inherits the condition when one of their two FBN1 genes mutates. The mutated gene dominates or overrides the normal copy. Below are three scenarios showing the autosomal dominant inheritance pattern.
Scenario #1: One Affected Parent (Most Common Scenario)
When one parent has Marfan syndrome, and the other does not, there is a 50% chance for each child to inherit the condition. Conversely, there is a 50% chance the child will not be affected.
- Affected parent: Has one mutated FBN1 gene (Dd).
- Unaffected parent: Has two normal FBN1 genes (dd).
- Outcome: Each child has a 50% chance of inheriting the mutated gene.
Scenario #2: Both Parents Are Affected (Rare Scenario)
When both parents have Marfan syndrome (MFS), there is a 75% chance the child will be affected (inheriting either one or two mutated genes). There is only a 25% chance the child will inherit normal genes from both parents and be unaffected.
Important note: Inheriting two mutated genes (DD) is very rare and typically results in much more severe symptoms than having only one mutated gene.
Scenario #3: De Novo Mutation (Spontaneous)
In about 25% of cases, neither parent carries the gene. Instead, a new (random) mutation occurs in the FBN1 gene during the formation of the egg or sperm, resulting in a child with Marfan syndrome despite no family history.
- Both parents have normal FBN1 genes.
- A spontaneous mutation occurs during conception.
- The child develops MFS without any family history.
- This child can then pass the mutated gene to their own children (50% chance).
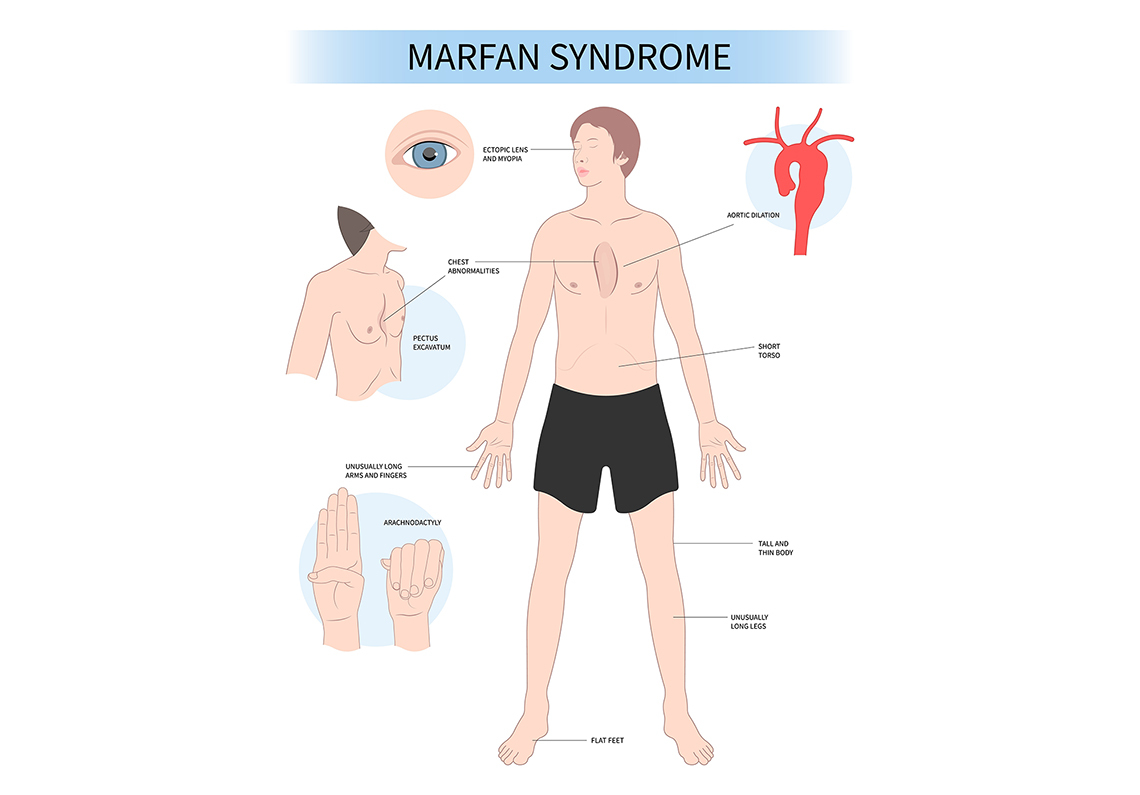
Diagnostic Features and Recognition
One of the diagnostic features that can help clinicians recognize Marfan syndrome is identifying characteristic facial features, often referred to as the Marfan syndrome face. These craniofacial features may be subtle, but when present together, they can aid in early recognition.
Clinical note: These features are more easily recognized in older children and adults. In infants and young children, facial features may be less pronounced.
- Dolichocephaly: Long, narrow face and skull.
- Downward-sloping palpebral fissures: Slanting of the eye openings.
- Enophthalmos: Deep-set eyes.
- Malar hypoplasia: Underdeveloped or flat cheekbones.
- High arched palate: Often associated with dental crowding.
- Retrognathia/micrognathia: Small or receding jaw (in some cases).
Distinctive body type characteristics include:
- Arm span exceeds height.
- Reduced upper-to-lower body segment ratio (legs are disproportionately long).
- Arachnodactyly (long, slender fingers and toes).
Chest wall deformities:
- Pectus excavatum (sunken chest) affects over 50% of individuals.
- Pectus carinatum (pigeon chest) - the breastbone protrudes outward.
These chest wall deformities often become more pronounced during adolescent growth spurts and can cause:
- Breathlessness and reduced exercise tolerance.
- Chest pain, particularly with exertion.
- Psychological distress and self-image concerns.
- Compression of the heart and lungs in severe cases.
Foot and joint abnormalities:
- Pes planus (flat feet) affects about 25% of patients.
- Increased risk of osteoarthritis at an early age.
- Protrusio acetabuli (hip socket abnormality where the femoral head pushes into the pelvis).
- Joint hypermobility (loose joints).
Cardiovascular Involvement in Marfan Syndrome
Medical issues related to the heart and blood vessels affect 9 out of every 10 people diagnosed with MFS. The connective tissue within the heart and blood vessels can be more flexible or fragile than usual. This can sometimes affect how the heart beats or how forcefully it pumps. While aortic issues (enlargement of the main artery) are often discussed, it is also important to understand the electrical and muscular changes that occur.
Aortic root dilation is the most common cardiovascular complication in people with Marfan syndrome. It happens in about 60-80% of individuals. It means the base of the aorta, the large blood vessel that carries blood from the heart, stretches and becomes larger than normal. When this area gets too big, the wall of the aorta becomes weak. This weakening is called an aortic aneurysm. As the aneurysm grows, the risk of the aorta tearing, called an aortic dissection, becomes much higher.
Warning signs of aortic dissection (a tear in the aorta):
- Sudden, severe chest pain (often described as tearing or ripping)
- Pain radiating to the back, abdomen, or neck
- Shortness of breath
- Loss of consciousness
- Symptoms of stroke (weakness, numbness) or reduced blood flow to limbs
Mitral valve prolapse occurs when the mitral valve becomes elongated and thickened, sometimes failing to close properly. Mitral valve prolapse affects 40 to 54% of people with Marfan syndrome, becomes more common with age, and occurs more often in women. Individuals with this condition may experience symptoms such as:
- Mitral valve regurgitation
- Heart palpitations
- Chest discomfort
- Fatigue
- Shortness of breath
- In severe cases, heart failure
Heart Muscle Function in Marfan Syndrome (Muscular Changes Seen in MFS)
Beyond the electrical changes seen in individuals with Marfan syndrome, the heart muscle can also have difficulty contracting. Reduced muscle strength can lead to problems. Two possible issues are outlined below.
- Mild left ventricular systolic dysfunction: The left ventricle is the hearts strongest chamber because it pumps oxygenrich blood to the entire body. In mild left ventricular systolic dysfunction, the heart is still able to pump blood out, but it does so in a slightly weakened state. This condition can usually be detected during an echocardiogram, and symptoms are often minimal or barely noticeable.
- Dilated cardiomyopathy: A less common but more serious condition is dilated cardiomyopathy. In this disorder, the hearts main pumping chamber becomes enlarged and weakened. As the chamber stretches, its ability to pump blood effectively decreases, which can lead to fatigue, shortness of breath, and other complications.
Ocular Manifestations of Marfan Syndrome
Marfan syndrome is associated with several eye-related findings that may affect vision and require ongoing monitoring.
Common Eye Problems
Ectopia lentis, or lens dislocation, is the most characteristic ocular feature of Marfan syndrome and occurs in approximately 5080% of affected individuals. It results from weakened zonular fibers that allow the lens to shift from its normal position and is typically identified during a comprehensive eye examination using a slit-lamp.
Strabismus and amblyopia may also be present. Strabismus refers to misalignment of the eyes, such as inward or outward turning, and if left untreated, may lead to amblyopia (lazy eye), in which the brain suppresses input from the weaker eye.
Refractive errors are common, particularly severe myopia, which occurs more frequently than in the general population. Some individuals may also have an enlarged corneal diameter, known as megalocornea.
Other potential ocular complications include:
- Increased risk of glaucoma due to elevated intraocular pressure.
- Higher risk of retinal detachment.
Pulmonary (Lung) Complications
Pulmonary involvement in Marfan syndrome most commonly presents as spontaneous pneumothorax, a condition in which air leaks into the space between the lung and chest wall, resulting in lung collapse. Current evidence suggests that pneumothorax occurs in approximately 411% of individuals with Marfan syndrome.
This complication may occur without trauma or strenuous activity and is often related to underlying abnormalities in lung tissue, such as blebs or bullae. Because spontaneous pneumothorax can cause acute respiratory symptoms and requires immediate medical attention, patient education on recognizing warning signs, including sudden chest pain and shortness of breath, is an important aspect of care.
Early Diagnosis, Intervention, and Prognosis
Early intervention means diagnosing Marfan syndrome as soon as possible and starting treatment right away. When doctors catch MFS early, they can monitor the heart and aorta closely, prescribe protective medications, and perform surgery before serious complications occur.
Dramatic Improvement in Survival
Historical perspective:
- Early 1970s: Average lifespan was only approximately 32 years (before modern interventions).
- By 1990: The Introduction of beta-blockers, regular aortic imaging, and preventive surgery had increased survival significantly.
- Present day: People diagnosed and managed appropriately can expect a life expectancy of nearly 70 years, with near-normal life expectancy when properly treated. Left untreated, the average life expectancy is 45 years.
Quality of Life
Modern medical care offers effective solutions for many complications:
- Surgical repair of chest deformities and scoliosis improves breathing and comfort.
- Treatment for lens dislocation restores clear vision.
- Medications help reduce symptoms and prevent life-threatening cardiovascular events.
- Life participation: Many people with MFS attend school, pursue careers, raise families, and stay physically active with only modest limitations.
Living with Marfan syndrome can present significant psychosocial challenges in addition to physical health concerns. Individuals may experience body image distress related to a tall, thin body habitus or visible skeletal features. Activity restrictions, particularly the need to avoid contact or high-intensity sports, can contribute to feelings of isolation or exclusion, especially among children and adolescents. Ongoing awareness of potential life-threatening complications, such as aortic dissection, may also lead to chronic anxiety or heightened health-related stress.
Diagnostic Criteria and Clinical Evaluation
Diagnosing MFS requires careful evaluation of multiple factors because features can vary widely even within families:
- Physical features (systemic score).
- Family history (genetic inheritance).
- Genetic testing (FBN1 mutation).
- Imaging results (cardiovascular and ocular).
The Ghent Nosology Criteria
It was originally created in 1996 and revised in 2010 (Ghent-2 criteria). It is the standardized clinical criteria for accurate diagnosis worldwide.
- Evaluates 7 diagnostic rules focusing on aortic root dilation and ectopia lentis as cardinal features.
- Applies to both inherited and de novo (new mutation) cases.
- Incorporates genetic testing (FBN1 mutation detection) as a major diagnostic criterion.
Systemic score: Ranges from 0 to 20 points. Measures skeletal and other physical features (wrist/thumb signs, chest deformity, etc.). A score of 7 points or higher indicates significant systemic involvement.
Imaging Tests
Specialized cardiovascular imaging includes:
- Transthoracic echocardiogram (TTE): Shows heart structure, valves, and proximal aorta (root). Standard initial test.
- Magnetic resonance angiogram (MRA): Displays all segments of the aorta throughout the chest and abdomen.
- Computed tomography (CT): Provides detailed images of all aorta segments; useful if magnetic resonance imaging (MRI) is contraindicated.
- Cardiac MRI: Helps determine timing for valve repair surgery and assesses ventricular function.
- Note: Plain X-rays and electrocardiogram (EKGs) are not sufficient for evaluating aortic root dimensions
Management and Treatment Strategies
Surgical Intervention:
- Elective aortic surgery is typically considered when the aortic root diameter reaches approximately 5.0 cm (or smaller with risk factors).
- Regular monitoring of aorta size is essential to time surgery appropriately.
- Early detection allows for timely repair (valve-sparing root replacement) before dissection or rupture.
Medical Management
Medications help slow disease progression and protect the cardiovascular system by reducing hemodynamic stress on the aorta.
Preferred medications:
- Beta-blockers: (e.g., atenolol, metoprolol). Carvedilol or metoprolol XL may be preferred for heart failure. They reduce heart rate and force of contraction, slow the rate of aortic dilation, and reduce stress on the aorta.
- Angiotensin Receptor Blockers (ARBs) (e.g., losartan, irbesartan): Recent meta-analysis research shows that ARBs significantly reduce the rate of aortic enlargement by about half, with or without beta-blockers. Often used if beta-blockers are not tolerated or in combination.
- Angiotensin-converting enzyme (ACE) inhibitors: Alternatives for blood pressure control and heart dysfunction.
Ocular Management
Diagnostic tools: Dilated eye examination, slit-lamp examination, eye pressure test (tonometry for glaucoma detection), visual acuity, and refraction testing.
Monitoring schedule: Annual eye exams (minimum) recommended for all patients. More frequent monitoring may be needed to determine if active problems exist.
Treatment options:
- Corrective lenses: Glasses or contacts for myopia/astigmatism.
- Medicated eye drops: To control eye pressure (glaucoma) or pupil size.
- Eye surgery: For significant lens dislocation (lensectomy), glaucoma, or retinal detachment.
- Important precautions:
- Avoid high-impact sports that could cause eye trauma (boxing, etc.).
- Wear protective polycarbonate eyeglasses during physical activity.
Orthopedic Management
Scoliosis correction:
- Considered for spinal curves of ≥ 45 degrees.
- Spinal fusion with instrumentation is the standard surgical treatment.
- Higher complication rates than the general population (cerebrospinal fluid [CSF] leaks, implant issues).
- Requires careful preoperative cardiac optimization.
Pectus repair (Ravitch or Nuss Procedure):
- Primarily for severe symptoms (compression of the heart/lungs) or significant cosmetic concerns.
- Conservative management (bracing) is often tried first for pectus carinatum.
Support Groups and Resources
Living with Marfan syndrome can be challenging, but you dont have to face it alone. Connecting with support groups and educational resources can provide valuable information, emotional support, and community connections for patients, families, and healthcare providers.
The Marfan Foundation
Website: www.marfan.org
Phone: 1-800-8-MARFAN (1-800-862-7326)
Services provided:
- Patient and family support programs.
- Educational materials and webinars.
- Annual family conferences.
- Research updates and clinical trial information.
- Marfan syndrome awareness campaigns.
- Healthcare provider resources and continuing education opportunities.
National Organization for Rare Disorders (NORD)
Website: www.rarediseases.org
Services: Comprehensive disease information, patient assistance programs, and advocacy resources.
Genetic and Rare Diseases Information Center (GARD)
Website: rarediseases.info.nih.gov
Services: Free information services from the National Institutes of Health for patients and families.
International Support Groups:
- Marfan Trust (UK): www.marfantrust.org
- European Marfan organizations: Country-specific support available through local registries.
About the Author:
Glenel Loring is a dedicated nurse transitioning into freelance health writing. With 20 years of bedside nursing experience, Glenel deeply understands healthcare. She is now looking to apply her medical expertise to craft engaging, informative content for diverse audiences. Glenel's writing highlights her ability to make complex health topics accessible and relatable. Her interest in herbal supplements as alternative health solutions showcases her dedication to lifelong learning. As a mother of two and avid gardener, Glenel brings a personal touch to her work. With her strong nursing background and emerging writing skills, Glenel seeks opportunities to help brands and publications educate and empower readers to take control of their well-being.
Glenel is an independent contributor to CEUfast's Nursing Blog Program. Please note that the views, thoughts, and opinions expressed in this blog post are solely those of the independent contributor and do not necessarily represent those of CEUfast. This blog post is not medical advice. Always consult with your personal healthcare provider for any health-related questions or concerns.
If you want to learn more about CEUfast's Nursing Blog Program or would like to submit a blog post for consideration, please visit https://ceufast.com/blog/submissions.